Publication
Article
Psychiatric Times
Rx Evolution: Pharmacological Paradigms for the Treatment of Schizophrenia
Author(s):
We remain challenged to improve treatments and outcomes in schizophrenia. Explore the latest options.


lily/AdobeStock

Much has been written about the stagnation of psychopharmacology for the treatment of schizophrenia. All US Food and Drug Administration (FDA)-approved medications for the treatment of the core symptoms of schizophrenia—psychosis, negative symptoms, and cognitive symptoms—are dopamine-2 (D2) receptor blockers, each of which has unique additional properties at other receptor systems.
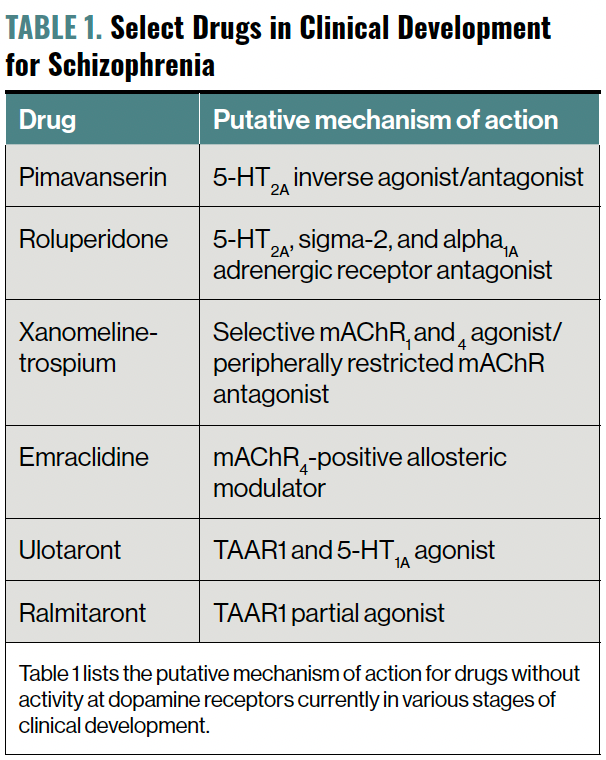
D2-blocking antipsychotics appear very close to finding themselves in the company of novel medications that treat schizophrenia by their action on non–D2 receptors, most notably as agonists at muscarinic acetylcholine receptors (mAChRs) 1 and 4, and the trace amine–associated receptor (TAAR) 1 (Table 1). Before exploring these novel mechanisms, let us review our current armamentarium for treating individuals with schizophrenia.


Table 1. Select Drugs in Clinical Development for Schizophrenia
The Birth of Antipsychotics
Chlorpromazine (Thorazine), a “dirty” molecule with activity at a wide range of neurotransmitter receptors, began this D2-blocker trajectory in the early 1950s. Initial attempts at improving efficacy and tolerability led to the development of D2 blockers that in retrospect were “too clean,” including haloperidol (Haldol) and fluphenazine (Prolixin). We learned from these selectively potent drugs that although they did treat psychotic symptoms, they worsened the negative and cognitive symptoms by blocking the D2 receptors in other important brain regions, as well as causing significantly more movement disorders and hyperprolactinemia. Significantly, over years and decades it is the negative and cognitive symptoms that contribute most to the progressive disability in many individuals with schizophrenia. Prior to chlorpromazine’s arrival in the United States, individuals with schizophrenia were often chronically institutionalized not for their positive symptoms, but for their negative and cognitive symptoms—hence the previous commonly used term dementia praecox (early-onset dementia).
Second-Generation Antipsychotics
Clozapine (Clozaril), initially developed in the 1950s, stood out as an effective antipsychotic for treatment-resistant patients, but its adoption in the United States was significantly delayed due to concerns of well-established severe neutropenia. Following reports of dramatic responses in some patients in other countries, clozapine was eventually FDA approved in 1989. The notable novel properties of clozapine included its activity at specific serotonin receptors, especially as a 5HT1A partial agonist and a 5HT2A antagonist. Clozapine remains our most effective antipsychotic and is the only drug FDA approved for treatment-resistant schizophrenia. Despite continuing research, we still do not understand the pharmacology that contributes to clozapine’s superior efficacy over other antipsychotics.
Continued research in an ongoing attempt to improve efficacy and tolerability led to 2 subclasses of antipsychotics: the “apines” (olanzapine, quetiapine, and asenapine, with structural similarity to clozapine) and the “odones” (risperidone, ziprasidone, paliperidone, iloperidone, and lurasidone), drugs that combined D2 blockade with 5HT2A antagonism and varying degrees of 5HT1A partial agonism. These second-generation antipsychotics continue to cause various movement disorders, albeit at about half the incidence and prevalence rates as the first generation.1,2
Additionally, some of these antipsychotics significantly increase the risk of weight gain, hyperglycemia or increased insulin levels, elevated lipids, sedation, and elevated prolactin.
Third-Generation Antipsychotics
In 2002, a novel class of antipsychotics arose with the FDA approval of aripiprazole (Abilify), a very potent D2 antagonist with intrinsic D2 partial agonism. When aripiprazole (A) is dosed to D2 receptor–antagonism saturation, the fixed amount of D2 partial agonism (based on the molecules’ physical chemistry properties) determines the maximum “functional” D2 antagonism that the brain would experience. Additional members of this family are brexpiprazole (B; Rexulti) and cariprazine (C; Vraylar). One of the shared properties of these “A, B, C” antipsychotics is that they do not raise prolactin levels as a direct result of their D2 receptor intrinsic partial agonism that prevents the postsynaptic intracellular environment from achieving more than 70% functional antagonism, a threshold required to raise prolactin at the tuberoinfundibular tract. Initial speculation that this D2 receptor–antagonism/partial agonism property would minimize or eliminate movement disorders resulting from their activity at the dorsal striatum proved to be incorrect, as akathisia and risk of tardive dyskinesia occur.
Finally, in 2019, lumateperone (Caplyta) was FDA approved for schizophrenia. This agent is functionally in a class of its own, with extremely potent 5HT2A antagonism and modest D2 antagonism, serotonin reuptake inhibition, and D1 receptor modulation.
Treating All Symptoms
Despite an impressive and heroic progression of research into the pathophysiology of schizophrenia, development of novel agents, and improvements in developing and delivering an accompanying essential array of biopsychosocial treatments and supports, the lifetime prognosis for many individuals with schizophrenia remains poor. Epidemiological data indicate that an individual with schizophrenia will die on average 20 years earlier than age-matched healthy individuals. Looking at the 1 dimension in which pharmacotherapy has had the greatest treatment results, psychotic symptoms, a staggering one-third of individuals with schizophrenia remain psychotic despite aggressive treatment. Another third continue with intermittent relapses of positive symptoms combined with progressive negative symptoms and cognitive decline. Currently there are no FDA-approved treatments to treat the negative and cognitive symptoms, which ultimately cause the greatest lifetime functional impairment.
As an exercise in curiosity, looking at our current understanding of the human brain through the eyes of a medication and psychiatric nosology–naïve basic neuroscientist, a reasonable and insightful assessment would be confusion as to why so much time, energy, and resources have remained focused on the 2 neurotransmitters dopamine and serotonin when there is so much more going on in our brains. To be fair, researchers have explored many alternative paths, but there has been no ultimate clinical advancement in treatments. It is likely that the dopamine stagnation will soon end. The current pipeline is bursting with fresh air. Let us explore the 3 novel mechanisms—all with no direct dopamine activity: drugs that continue to bind to serotonin receptors; drugs that agonize central mAChRs; and drugs that agonize the novel TAAR 1.
Serotonin Activity – 5HT2A
Pimavanserin. Currently, pimavanserin (Nuplazid) is only FDA approved to treat hallucinations and delusions associated with Parkinson disease psychosis, for which it received approval in 2016. Significantly, it lacks any activity at dopamine receptors and its primary mechanism of action is believed to be related to its potent 5-HT2A inverse agonist/antagonist activity. In theory, it is possible that increased antagonism at the 5-HT2A receptor may improve negative symptoms, as well as possibly enhance the antipsychotic effect as an augmentation agent to a primary antipsychotic medication. It is well established that the 3 hallucinogens psilocybin, mescaline, and LSD all share the primary mechanism of 5-HT2A agonism. This fact suggests that the 5-HT2A receptor may somehow contribute to psychotic symptoms in schizophrenia.
Two studies were designed to explore these potential clinical benefits. The ADVANCE trial was a double-blind, placebo-controlled, randomized 26-week study adding pimavanserin as augmentation to the primary antipsychotic agent (most common were risperidone, olanzapine, and aripiprazole) in stable outpatients with schizophrenia who demonstrated significant negative symptoms. The primary outcome was change from baseline in the Negative Symptom Assessment-16. At study end, pimavanserin demonstrated a –10.4-point reduction versus a –8.5- point reduction in placebo (P = .043; effect size = 0.21). Although a slight improvement was observed in the pimavanserin cohort, an effect size of 0.21 is weak.
A second study, the ENHANCE-1 trial, looked at stable outpatients with schizophrenia who had an inadequate response to antipsychotics with ongoing positive symptoms. It was a placebo-controlled, 6-week, randomized, double-blind trial of adjunctive pimavanserin. At study end, 56.5% of patients who received pimavanserin versus 50.5% of patients who received placebo demonstrated a 20% reduction in their Positive and Negative Syndrome Scale (PANSS) score. Hence, collectively, the data from these 2 trials demonstrated minimal clinical advantage to antipsychotic augmentation with pimavanserin.3
Roluperidone. Roluperidone is a novel molecule that has antagonism at the 5-HT2A, sigma2, and alpha1A adrenergic receptors. It has been investigated in two 12-week randomized, double-blind, placebo-controlled trials in individuals with schizophrenia who have been symptomatically stable but demonstrated significant negative symptoms with a PANSS negative symptoms subscale greater than 20. The primary end point was the PANSS-derived Negative Symptom Factor Score (NSFS). A secondary end point was the Personal and Social Performance scale total score. All other psychotropic medications were discontinued 2 days prior to randomization and throughout the study. The only exception was the as-needed use of the rescue medications lorazepam (Ativan) for agitation and zolpidem (Ambien) for insomnia, and an anticholinergic for emergent EPS.
Data from both studies demonstrated an improvement in the 64-mg roluperidone dose compared with placebo in the primary and secondary outcomes. Additional studies are ongoing. Roluperidone shares the antagonism at 5-HT2A with pimavanserin and most of the second-generation antipsychotics, and antagonism of alpha1A adrenergic receptor with many of the existing antipsychotics. The function of sigma2 receptors is poorly understood. Although the sigma2 receptor was differentiated as a unique receptor from sigma1 in 1990, the gene coding for this receptor was not identified until 2017.4
Muscarinic Cholinergic Agonism
Acetylcholine is believed to be the first neurotransmitter, being synthesized from its precursor choline, which was used billions of years ago for cell membrane synthesis by single-celled organisms. It is well established that acetylcholine served a functional role in primitive organisms that lacked a nervous system. The 2 major acetylcholine receptor subfamilies—nicotinic and muscarinic, both ubiquitous in living systems—evolved independently. Nicotinic cholinergic receptors, which are donut-shaped pentamers with numerous subunits, function as ligand-gated ion channels and evolved from a common ancestor receptor over 2.5 billion years ago. mAChRs are metabotropic G protein–coupled receptors with 5 subfamilies (M1 through M5) and diverged from the G protein–coupled receptor super-family of receptors between 0.5 and 1 billion years ago. Not surprisingly, acetylcholine is a foundational neurotransmitter in humans, with critical functions throughout the brain and body.5
Research dating back to 1957 demonstrated that the naturally occurring mAChR agonist arecoline, when given to patients with schizophrenia, resulted in “lucid intervals.”6 Notably, arecoline occurs naturally in the betel nut, which is chewed in some Asian cultures. Findings from limited studies have shown that individuals with schizophrenia demonstrated an improvement in both positive and negative symptoms following betel nut chewing.7
In the 1990s, a synthetic structural analogue of arecoline, xanomeline, was characterized and ultimately studied in patients with Alzheimer disease. It demonstrated improvement in cognition, as well as a decrease in psychotic symptoms when present in these patients. This led to a subsequent study in individuals with treatment-resistant schizophrenia in which antipsychotic and improved cognitive function were observed. Poor tolerability resulting from the agonism of peripheral mAChRs ultimately led to the temporary abandonment of further research in this population. Monotherapy with xanomeline resulted in adverse effects, including nausea, vomiting, diarrhea, sweating, and hypersalivation.
To address these unacceptable adverse effects, Karuna Therapeutics hypothesized and confirmed clinically that with the addition of trospium—a well-established peripherally restricted mAChR antagonist as a combination molecule—to xanomeline, the central benefits of mAChRs’ agonism could be maintained, as trospium dampens the adverse effects by antagonizing the peripheral mAChRs. The resulting combination drug, xanomeline-trospium (KarXT), was compared with placebo in a phase 2 double-blind trial in individuals with schizophrenia for 5 weeks. Investigators enrolled 182 participants with an average PANSS score of 97. At 5 weeks, the PANSS score in the xanomeline-trospium group improved –17.4 points in comparison with an improvement of –5.9 points in the placebo group (P < .001). The most common adverse effects in the active treatment group included constipation, nausea, dry mouth, dyspepsia, and vomiting.8 These phase 2 results were recently replicated in a phase 3 trial.9
The past 2 decades have resulted in a wealth of increasingly complex information that has revealed important functional differences in the neurophysiology of the 5 mAChRs. Central M1 and M4 receptor agonism is associated with improvements in cognition and psychosis. This finding is consistent with the early research that demonstrated an improvement in both cognition and psychosis in patients with Alzheimer disease and schizophrenia with xanomeline, a synthetic agonist with selectivity at M1 and M4 receptors. The interested reader is referred to an excellent and comprehensive review of the mAChR agonist story that was recently published.10
A second mAChR agonist in development for the treatment of schizophrenia by Cerevel Therapeutics is the M4-positive allosteric modulator emraclidine (CVL-231). It is highly specific for M4 and lacks any appreciable agonism at M1, hence negating the need to simultaneously block peripheral M1 receptors. In a recent phase 1b trial in individuals with schizophrenia, emraclidine produced clinically meaningful and statistically significant improvement at week 6 compared with placebo in the PANSS total score. Currently, two phase 2 trials are underway.11
Trace Amine–Associated Receptor 1
Trace amines were characterized in the 1970s, and the original definition was any endogenous monoamine present in physiological levels at least 2 orders of magnitude lower than serotonin, dopamine, and norepinephrine. The most prominent trace amines are β-phenylethylamine, p-tyramine, tryptamine, and p-octopamine. Initial hypotheses of their function were dead ends, and they were all but forgotten until the discovery of TAARs by 2 separate groups in 2001. This novel class of G protein–coupled receptors includes at least 26 subtypes discovered in vertebrates to date, with 6 functional members confirmed in humans (TAAR1, 2, 5, 6, 8, and 9).
In humans, TAAR1 is the most prevalent and diverse TAAR. It is found in brain structures involved in psychosis, reward, mood, anxiety, and cognition. Outside of the central nervous system, TAAR1 receptors are located in the stomach, intestines, pancreatic beta cells, and leukocytes.12,13 Research to date has established that TAAR1 regulates dopamine neurotransmission to keep dopamine concentrations within an ideal physiological range. TAAR1 receptor agonism decreases dopamine presynaptic release, hence preventing a hyperdopaminergic state, whereas TAAR1 antagonism increases dopamine release in hypodopaminergic states. This is the putative mechanism by which psychotic symptoms may be decreased.
TAAR1 is present intracellularly both pre- and postsynaptic. It appears that when activated, TAAR1 forms a heterodimer with D2 receptors and may flip this complex across the membrane. Hence TAAR1 appears to function as an endogenous rheostat—maintaining neurotransmitter concentrations to maximize homeostasis. In addition to dopamine, TAAR1 regulates glutamate concentrations, which appears to prevent hypoglutamatergic states in the prefrontal cortex that could explain its procognitive effects. Finally, TAAR1 is active in the serotonin system as well, with expression in the dorsal raphe nucleus.
Beyond demonstrated benefits in psychotic, cognitive, and negative symptoms in schizophrenia, preclinical evidence suggests TAAR1 modulation may also prove beneficial in the treatment of mood and anxiety disorders, sleep disorders, metabolic syndrome, obesity, and substance use disorders.
Ulotaront is a TAAR1 and 5-HT1A agonist currently in phase 3 studies for the treatment of schizophrenia. In a completed 4-week, phase 2, double-blind, placebo-controlled, randomized study in adults with schizophrenia in an acute psychotic relapse, the agent demonstrated significant improvement compared with placebo. A subset of these participants was treated in an open-label 26-week extension study, and showed continued improvement (Table 2).


Table 2. Improvement in Mean PANSS Scores
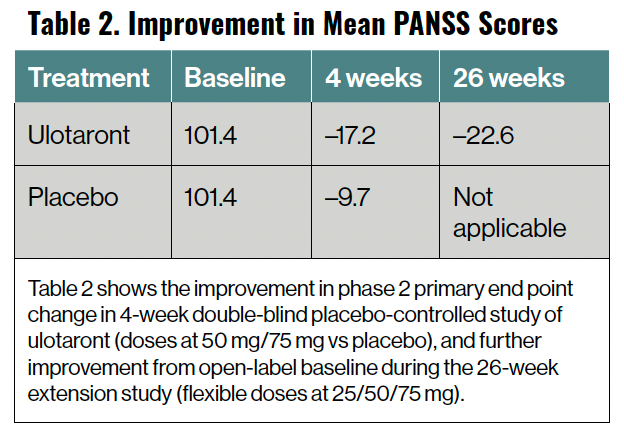
In data from both the 4-week phase 2 study and the open-label 26-week extension study, the tolerability and safety of ulotaront was similar to that of placebo. Ulotaront demonstrated no significant effect on movement disorders, lipids, glycemic indices, weight, electrocardiogram changes, or prolactin. In the 4-week phase 2 study, diarrhea and dyspepsia were the only adverse effects that occurred at a rate greater than 2% and at twice the rate of those of placebo. Phase 3 studies are well underway.14,15
A second TAAR1 agent, ralmitaront, functions as a partial agonist and is being studied in a phase 2 trial in schizophrenia.16
Concluding Thoughts
The understanding and treatment of schizophrenia over the past 70 years has been fruitful on many fronts. Significant progress has been made in the understanding of the interplay of numerous risk factors through wide-ranging research, from genetics and epigenetics to developmental and environmental factors, neuroimaging, and more.
However, as is the case with many chronic disorders such as cardiovascular disease and diabetes, we remain challenged to improve our treatments and outcomes. Treating all 3 domains of schizophrenia—positive, negative, and cognitive symptoms—continues to be a high priority and significant unmet need. Three novel classes of drugs—5-HT2A receptor antagonists, mAChR 1 and 4 agonists, and TAAR1 agonists—may write the next chapter on non–D2 receptor drug treatments. These treatments may improve the cognitive and negative symptoms as well as the positive, possibly as monotherapies and possibly as combination treatments with traditional antipsychotics.
Dr Miller is Medical Director, Brain Health, Exeter, New Hampshire; Editor in Chief, Psychiatric Times™; Staff Psychiatrist, Seacoast Mental Health Center, Exeter; Consulting Psychiatrist, Exeter Hospital, Exeter; Consulting Psychiatrist, Insight Meditation Society, Barre, Massachusetts.
References
1. Carbon M, Hsieh CH, Kane JM, Correll CU. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278.
2. Carbon M, Kane JM, Leucht S, Correll CU. Tardive dyskinesia risk with first- and second-generation antipsychotics in comparative randomized controlled trials: a meta-analysis. World Psychiatry. 2018;17(3):330-340.
3. Davis J, Zamora D, Horowitz M, Leucht S. Evaluating pimavanserin as a treatment for psychiatric disorders: a pharmacological property in search of an indication. Expert Opin Pharmacother. 2021;22(13):1651-1660.
4. Davidson M, Saoud J, Staner C, et al. Efficacy and safety of roluperidone for the treatment of negative symptoms of schizophrenia. Schizophr Bull. 2022;48(3):609-619.
5. Dean B. Evolution of the human CNS cholineric system: has this resulted in the emergence of psychiatric disease? Aust N Z J Psychiatry. 2009;43(11):1016-1028.
6. Pfeiffer CC, Jenney EH. The inhibition of the conditioned response and the counteraction of schizophrenia by muscarinic stimulation of the brain. Ann N Y Acad Sci. 1957;66(3):753-764.
7. Sullivan RJ, Allen JS, Otto C, et al. Effects of chewing betel nut (Areca catechu) on the symptoms of people with schizophrenia in Palau, Micronesia. Br J Psychiatry. 2000;177:174-178.
8. Brannan SK, Sawchak S, Miller AC, et al. Muscarinic cholinergic receptor agonist and peripheral antagonist for schizophrenia. N Engl J Med. 2021;384(8):717-726.
9. Correll CU, Angelov AS, Brannan SK. Safety and efficacy of KarXT (xanomeline-trospium) in patients with schizophrenia: results from a phase 3, randomised, double-blind, placebo-controlled trial (EMERGENT-2). Karuna Therapeutics. Accessed December 7, 2022. https://karunatx.com/wp-content/uploads/2022/10/ECNP-2022_Poster_EMERGENT-2_Correll_2022-10-16_FINAL_.pdf
10. Paul SM, Yohn SE, Popiolek M, et al. Muscarinic acetylcholine receptor agonists as novel treatments for schizophrenia. Am J Psychiatry. 2022;179(9):611-627.
11. Krystal JH, Kane JM, Correll CU, et al. CVL-231 as a novel positive allosteric modulator targeting M4 muscarinic receptors: results from a phase 1b trial in patients with schizophrenia. Cerevel Therapeutics. Accessed December 7, 2022. https://investors.cerevel.com/static-files/d8c8d2c7-3689-4be9-978a-c6b5a331be4a
12. Berry MD, Gainetdinov RR, Hoener MC, Shahid M. Pharmacology of human trace amine-associated receptors: therapeutic opportunities and challenges. Pharmacol Ther. 2017;180:161-180.
13. Dedic N, Dworak H, Zeni C, et al. Therapeutic potential of TAAR1 agonists in schizophrenia: evidence from preclinical models and clinical studies. Int J Mol Sci. 2021;22(24):13185.
14. Koblan KS, Kent J, Hopkins SC, et al. A non–D2-receptor-binding drug for the treatment of schizophrenia. N Engl J Med. 2020;382(16):1497-1506.
15. Correll CU, Koblan KS, Hopkins SC, et al. Safety and effectiveness of ulotaront (SEP-363856) in schizophrenia: results of a 6-month, open-label extension study. NPJ Schizophr. 2021;7(1):63.
16. Kantrowitz JT. Trace amine-associated receptor 1 as a target for the development of new antipsychotics: current status of research and future directions. CNS Drugs. 2021;35(11):1153-1161.















Newsletter
Receive trusted psychiatric news, expert analysis, and clinical insights — subscribe today to support your practice and your patients.